
Welcome to the Paragraph Four Report®. Here, we bring some clarity to the confusion surrounding the world of generic drug approval process, Paragraph IV certifications, and patent challenges. There is an ever-increasing degree of activity in this area which means that things have gotten a bit more difficult to understand.
As with most things we do at The Paragraph Four Report, we are keeping this section simple. While it discusses the rules and reasons why and it remains accurate, there may be the exception to the exception that we won’t explore. But you will get the idea.
Though you can click on any of the following topics to begin, we suggest that you go in order if you are new to all of this.
The Basics
The Food and Drug Administration (FDA) oversees the drug approval process in the U.S. When branded drug companies such as Pfizer or GlaxoSmithKline (also referred to as “manufacturers”), want to get a new drug approved, they must provide data to the FDA showing that the drug is both safe and effective. The manufacturers provide the data in a New Drug Application (NDA) where they place all of the scientific and clinical data, usually from at least two well-controlled clinical studies.
If approved, the drug will have two forms of market protection. The first comes in the form of “exclusivity” which is a creation of law. Exclusivity enables the drug product to have exclusive, or monopoly, status in the market for a certain number of years (five years for a new chemical entity and other periods of time for different situations.) Exclusivity means that the FDA cannot legally approve a generic drug application for that product until the exclusivity period expires.
Exclusivity should not be confused with patent protection, which is the second form of market protection for a marketed branded drug. Included in the NDA is information regarding any patents on the drug product. The manufacturer applies for these patents with the US Patent and Trademark Office which, of course, works independently from the FDA. The FDA lists the patents in the Approved Drug Products with Therapeutic Equivalence Evaluations, also known as the Orange Book.
When a generic company wants to sell its generic version of a branded product, it usually does so through the Abbreviated New Drug Application (ANDA). When it files an ANDA, it must show that it meets certain bioequivalence standards and must certify against any listed patents. The generic approval process is discussed in more detail in the next section.
The Generic Approval Process
In 1984, Congress passed The Drug Price Competition and Patent Term Restoration Act that came to be known as the “Hatch-Waxman Act.” The Hatch-Waxman rules created processes and incentives for both branded and generic companies involving challenges to patents.
- Branded pharmaceutical companies are required to list patents involving composition of matter (substance), formulation, and method of use in the Food and Drug Administration’s (FDA) Approved Drug Products with Therapeutic Equivalence Evaluations, also known as the Orange Book.
- When applying to enter the market with a generic form of a reference product, the generic company files an Abbreviated New Drug Application (ANDA) and certifies against patents listed in the Orange Book. The certification states that either (a) FDA should approve of its generic version after the date the last patent expires (a “Paragraph 3” filing) or (b) that its generic product does not infringe on the listed patents or that those patents are not enforceable (a “Paragraph 4” filing actually called a Paragraph IV filing.)1
- If the generic company files an ANDA with a Paragraph IV certification, then the branded company is notified. After the notice, the branded company has 45 days to file a patent infringement action against the generic company. After the suit has been filed, the FDA cannot approve of the application until the generic company successfully defends the suit or until 30 months, whichever comes first.
1 The Regulations also allow for two other types of certifications that are not relevant for The Paragraph Four Report. A Paragraph 1 certification states that there are no patents listed, and a Paragraph 2 certification states that the listed patents have expired.
Why Generic Companies Challenge Patents
An important section of Hatch-Waxman Act actually encourages generic companies to challenge patents. If a generic company is the first to file its Abbreviated New Drug Application (ANDA) with a Paragraph IV certification and prevails in the subsequent lawsuit, that generic company is granted a period of market exclusivity of 180 days.
The 180-day exclusivity incentive can be significant for a generic company as it would be the only generic version on the market. So, it can price its product slightly below the branded version for six months, take market share from the branded product, and maintain its price point before other generics enter the market and erode the price and segment margins. The additional profit for a generic firm can be enormous if the product it challenges is a so-called blockbuster or megabrand.
The following example best shows how this works and why a generic company would go through this process.
Generic company Barr Laboratories identified Lilly’s Prozac®(fluoxetine) as a product with a large market potential and a patent position that could be challenged. In 1996, Barr filed its ANDA with a Paragraph IV certification, claiming that its version of fluoxetine did not infringe on the Lilly patents or that the patents were unenforceable.After five years of litigation, on August 9, 2000, the Court of Appeals vacated a lower court decision, ruling in favor of Barr and invalidating one of the patents for Prozac. After the compound patent expired and a pediatric exclusivity expired Prozac on August 2, 2001, Barr launched its generic form of the 20mg strength of fluoxetine which alone took 65% of the Prozac share in the first two months after it launched.
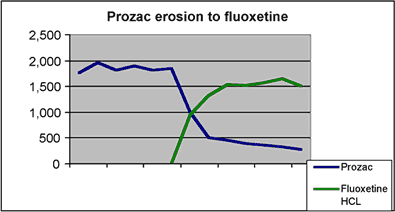
This graph shows the dramatic uptake of generic fluoxetine. Within the first month of the introduction of the Barr form of fluoxetine, Prozac lost 46% of its total prescriptions.2 By the end of the six month time period of exclusivity for Barr, Prozac had lost 82% of its prescriptions to the generic form and held onto only 16% of the Prozac/fluoxetine market. 3 The Hatch-Waxman incentives enabled Barr to successfully challenge the fluoxetine patent and reap the economic benefits of its own exclusivity period. In the eleven months after launch, the sales of Barr’s generic fluoxetine reached $367.5M which accounted for 31% of the company’s total product sales.
After other generic companies entered the market with their forms of fluoxetine and eroded the price, the company estimated that the sales of its form of fluoxetine decreased to 1% of its total product sales. 4When comparing profit margins generated by the fluoxetine case, it becomes quite obvious why Paragraph IV filings are part of its core business strategy. During the last quarter of 2000, Barr reported Product Sales of $142M and a gross margin (deducting the Costs of Sales and Selling & Administration Costs) of $24M, leaving a gross profit margin of 16.8%.However, during the last quarter of 2001, when it had a monopoly on generic fluoxetine sales, Barr took in $360M in Product Sales. With a gross margin of $103.9M, Barr increase its gross profit margin to 28.7%. 5 Hence, by succeeding with its Paragraph IV challenge, it nearly doubled its gross profit margin.
In addition, Barr also had a dramatic increase in its stock price. During the month of July 2001, when the favorable Court decision was rendered, Barr’s stock price was at a low of $66.00. By the end of the month, it had spiked to $89.38, realizing a gain of over 35%.
The successful Barr challenge of Prozac is often-cited as the case which first created a great deal of interest for ANDA filers to challenge patents under a PIV certification. Over time, this proved to be true. While this example is somewhat old and historic, the principles remain the same — being ‘first-to-file” ANDA is a powerful financial incentive and generic erosion of brand sales became even more pronounced since this case.
1 Drug Digest
4,5 Barr SEC 10-K filing for period ending December 31, 2001
An Emerging Trend
Indeed, most, if not all, generic companies have made the Paragraph IV certification a routine part of doing business. Since 2000, many companies have entered the PIV Market, and there has been a steady uptick in PIV cases (per ParagraphFour.com research). For example, in:
2005 — 77 PIV cases were filed
2010 — 234 cases were filed and
2015 — 402 cases were filed
The PIV certification has arguably become the most viable pathway for an ANDA filer to reach the generic market. Without filing a PIV certification, an ANDA filer can find itself reaching the market long after the product’s price has eroded too low to ensure any reasonable profit.
Size of Brand is No Barrier and When PIV Certifications are Filed is Often Unpredictable
As mentioned in the prior section, the Barr victory in the blockbuster Prozac®(fluoxetine) case was an important starting point for the Emerging Trend. However, the size of the brand is not the only factor in determining whether it will receive a PIV challenge. In fact, brands of all levels of sales have received PIV certifications over the years.
Moreover, when a brand receives a PIV certification can be unpredictable as well. Since 2000, some brands have received a PIV certification within months of approval but at other times it may be years before it receives its first PIV certification. The only exception to this general observation is for NCE products which have certain rules around when they can first receive a PIV certification (four years after approval). Since about 2012, many of these “NCE-1” products have received multiple ANDA filers at year four.
However, while intuition might suggest that: (1) nearly every brand receives a PIV certification; (2) there are always many (4+) ANDA PIV filers per product; and (3) there are always many (4+) ANDA filers at time of generic launch, the data do not support these assertions. In fact, in research compiled for ParagraphFour.com subscribers, the data often show the opposite.
Inter Partes Reviews, Petitions, and PIV Cases
In many instances, there are correlations between pending PIV cases and citizens petitions filed with the FDA as well as PIV cases and petitions filed for Inter Partes Review with the USPTO. The IPR Process began in 2011 and enables a different way to challenge a patent. IPR proceedings will run in parallel to PIV cases and at times judges presiding over PIV cases will defer to the IPR proceeding. Petitions can also affect the product that is under PIV certification in a variety of ways.